
CorrectSequence Therapeutics (Correctseq), a biotechnology company incubated from ShanghaiTech using innovative base editing technology to help people with severe diseases, today announced that the company and the research teams from ShanghaiTech University, Wuhan University, Fudan University and Children’s Hospital of Fudan University published an article entitled “Base editing of the HBG promoter induces potent fetal hemoglobin expression with no detectable off-target mutations in human HSCs” in Cell Stem Cell on November 20th, reporting a novel base editing strategy for treating β-hemoglobinopathies with transformer base editor (tBE). Associate Professor Chen Jia in SLST and Assistant Professor Yang Bei in SIAIS & SLST are two of the four corresponding authors.
Compared with Cas nuclease-mediated gene editing therapy, tBE editing exhibited higher γ-globin induction, lower cytotoxicity and undetected off-target mutations. Up to now, the pipeline for β-hemoglobinopathies developed by Correctseq based on tBE has exhibited long-term safety and efficacy in mice for more than 6 months and will enter clinical trials by the end of the year. With in-depth understanding in the therapeutic mechanisms of β-hemoglobinopathies using various genome editors at multiple targets, the pipeline under development in Correctseq with tBE has demonstrated great potential as the best-in-class gene editing therapy for the patients around the world.
β-hemoglobinopathies, including β-thalassemia and sickle cell disease (SCD), are the severe human autosomal monogenic genetic diseases caused by the mutations in hemoglobin subunit beta (HBB) gene locus. More than 3% of the global population carries the beta-hemoglobinopathy-associated alleles, and hundreds of thousands of newborns are diagnosed each year.
Reactivating silenced γ-globin expression offers a universal therapeutic strategy for treating β-hemoglobinopathies. γ-Globin expression is tightly regulated during development and is silenced shortly after birth by the repressors that bind to HBG1/2 promoter regions. BCL11A and ZBTB7A are the two major repressors of γ-globin gene expression, responsible for its silencing.
A therapeutic strategy disrupts the erythroid-specific BCL11A enhancer located on chromosome 2 via CRISPR/Cas9 greatly reduces BCL11A expression in erythroid cells without affecting other lineage development. Using Cas9 nuclease to disturb repressor binding motifs in hematopoietic stem and progenitor cells (HSPCs) relies on generating DSBs, thereby raising the risk of p53-dependent DNA damage response and cell toxicity in HSPCs. Moreover, the studies about β-thalassemia patients with naturally reactivated γ-globin expression have indicated that the fetal hemoglobin (HbF, α2γ2) level correlates negatively with the number of morbidities. Therefore, it is important to develop a safe and efficacious therapeutic strategy for β-hemoglobinopathies, which efficiently promotes HbF expression at a level sufficient for cure, meanwhile circumventing safety issues such as DSBs and off-target mutations.
At first, the researchers designed pairs of sgRNAs and helper sgRNAs targeting six major regulatory motifs in the regulatory network of HBG1/2 (Figure 1, top box). After examining on-target and gRNA-dependent off-target editing with five other previously published CBEs, the authors found tBE displayed no detectable off-target mutations across all six sgRNAs, as well as high on-target editing efficiency. Then, the researchers optimized the RNA expression system of tBE and electroporation conditions, which greatly improved the editing efficiency of tBE in HSPCs. To evaluate the therapeutic potential of tBE-based editing strategies, they electroporated mRNA encoding tBE and chemically modified sgRNA/hsgRNA pairs into the HUDEP-2 cell line, and measured the reactivated γ-globin mRNA expression. the authors found that editing the BCL11A binding motif located at the HBG1/2 promoter produced the highest γ-globin mRNA levels, which was chosen as the therapeutic target for tBE-editing strategy. The authors further compared tBE-editing strategy with other preclinical and clinical method, including the therapeutic sgRNA targeting BCL11A erythroid enhancer that has been approved to treat sickle cell disease—sgBCL11A-1617 (Figure 1, bottom left box). The tBE-edited HSPCs were then in vitro differentiated into erythroid cells to measure the γ-globin mRNA and protein expression levels, and the authors found that tBE-editing strategy produced significantly more γ-globin mRNA and protein than other gene editing strategies.
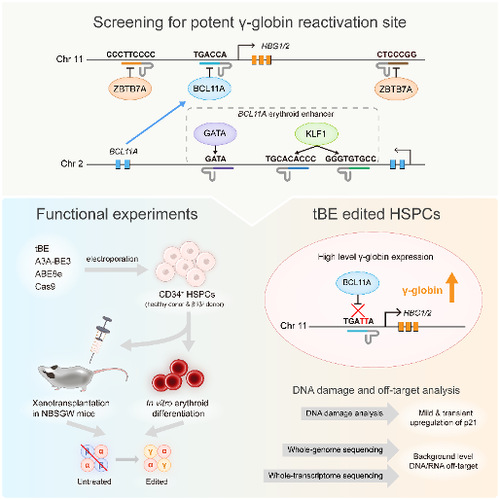
Figure 1. Schematic diagram of the main steps of this study
To determine whether tBE-edited HSPCs retained repopulation ability and whether the editing persisted over the differentiation process, the researchers xenotransplanted tBE-edited HSPCs into immunodeficient mice (Figure 1, bottom left box). No significant differences were observed between the edited and the mock-treated HSPCs in terms of engraftment and differentiation potential after 4 months, which suggests that the edited alleles were maintained in hematopoietic stem cells (HSCs) and in their progenies.
Furthermore, the authors compared the efficacies of different gene editing strategies to treat CD34+ HSPCs from β-thalassemia patient and found that the editing of the BCL11A binding site at HBG1/2 promoter by tBE_sgHBG-1 exhibited the highest γ-globin reactivation. At the same time, the research team conducted a comprehensive assessment of the level of DNA damage and DNA/RNA off-target events during tBE editing of HSPCs. The experimental results showed that tBE-edited HSPCs only transiently and slightly up-regulated the expression level of p21, and no DNA/RNA off-target mutations higher than the background level were detected (Figure 1, bottom right).
Collectively, tBE-mediated editing at the BCL11A binding site in the HBG1/2 promoter triggers robust induction of HbF with no detectable off-target mutation, demonstrating the potential to become the “Best in Class” approach for the treatment of β-hemoglobinopathy in clinics.
*This article is provided by Correctseq